
Osteogenesis Imperfecta
Clinical Features of Type I Osteogenesis Imperfecta
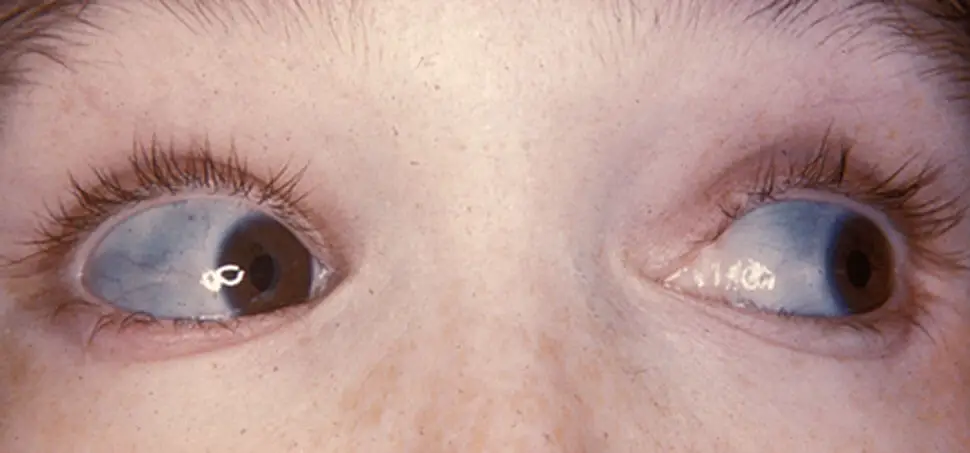 Figure X-1, Characteristically blue sclera of patient with osteogenesis imperfecta | Image courtesy of Wikipedia Opens in new window
Figure X-1, Characteristically blue sclera of patient with osteogenesis imperfecta | Image courtesy of Wikipedia Opens in new window
Osteogenesis imperfecta type I is the most common and mildest form of osteogenesis imperfecta Opens in new window.
This form is autosomal dominantly inherited and is characterized by mild bone fragility, with fractures occurring after moderate trauma, and by blue sclera and conductive hearing loss. It is subdivided into A and B based on the presence of dentinogenesis imperfecta Opens in new window.
The clinical signs and symptoms of both subtypes include blue sclera, in utero fractures in 10% of patients (fractures are more common during infancy), mild-to-moderate bone fragility with frequency of fractures decreasing after puberty, kyphoscoliosis, hearing loss, easy bruising, and short stature.
Findings of these clinical features are discussed in the entry under the following headings:
- Ophthalmologic abnormalities
A hallmark of this syndrome is blue sclera. It was noted early (5) that the severity of osteogenesis imperfecta was different depending on the presence or absence of blue sclera Opens in new window.
Scleral color appears to be consistent within families, although the degree of blueness varies from one family to another (10). Sclera blueness is believed to arise from disordered molecular organization (9).
The cornea may appear thinner than usual by slit-lamp examination (11). Embryotoxon Opens in new window has also been noted (14). It is unknown whether this abnormality is the result of chance association of hypercholesterolemia or whether it is one of the pleiotropic effects of the syndrome, at least in some families. Rarely, retinal detachment occurs (13).
- Otolaryngologic abnormalities
Hearing loss, rarely detected before 10 years of age, usually begins with a conductive deficit in the late second or early third decade (15).
With age, mixed and sensorineural hearing losses are observed (15, 16), the sensorineural component being the major one.
Reidner et al (15) and Garretsen and Cremers (17) noted that by the fifth decade, half of all patients had hearing loss, whereas by the seventh decade, all individuals had hearing loss.
Shapiro et al reported audiologic abnormalities in a heterogeneous group of patients over 30 had hearing loss. Half of all patients examined had sensorineural loss.
Conductive loss in this syndrome has been attributed to ossicular immobility at the stapes footplate (15). Fracture of the stapedial crura and atrophy of the stapes may also contribute to loss of hearing acuity (15).
- Cardiovascular involvement
The frequency of symptomatic cardiovascular anomalies is low. Hortop et al (18) reported nonprogressive aortic root dilatation in about 12% of affected patients.
In one study of dominant osteogenesis imperfecta, 9% of males and females had asymptomatc mitral valve prolapse Opens in new window; 24% of males but only 4% of females had asymptomatic aortic root dilatation (19).
Aortic regurgitation has been observed in patients after the third decade, as has mitral regurgitation. Aortic aneurysm and dissection do not occur, although left atrial rupture has been reported in one patient.
Mitral valve leaflets were thin in half the patients reported by White et al (20). They may cause mitral valve prolapsed and regurgitation. Microscopic findings in the valves include myxoid degeneration and atrophy and, in the aorta, cystic medial necrosis.
- Neurologic manifestations
Results of computed tomography (CT) scans of the head have been normal in the few patients tested, and ventricles are normal in size.
In the three-member family with osteogenesis imperfecta type 1 and dental abnormalities reported by Pozo et al (27), advanced basilar impression (platybasia Opens in new window) resulted in ventricular dilatation, multiple neurological disturbances of the foramen magnum compression syndrome, and death from acute brain stem compression.
These findings, however, are likely rare in other patients with this syndrome. These investigators noted that all patients reported with osteogenesis imperfecta and basilar impression had mild skeletal disease.
Rarely, radicular compression, trigeminal neuralgia, and communicating hydrocephaly occur. Half the 56 patients with osteogenesis imperfecta studied by Reite and Solomons had abnormal electroencephalograms; however, their patients were a heterogeneous group and were not classified.
- Joint abnormalities
Joint hypermobility and joint dislocation are found (21). Separate entities with either a phenotype resembling Ehlers-Danlos syndrome Opens in new window or with rigid joints or contractures (Bruck syndrome) exist.
- Skeletal manifestations
Macrocephaly Opens in new window has been reported in this syndrome, of which head size is usually large for height: the median of the distribution of head sizes is above the 50th centile (2).
Wormian bones Opens in new window may be present (2). Platybasia and occipitalization of the upper cervical vertebrae may produce a “tam o’shanter” appearance.
Multiple fractures usually occur, although about 10% of patients may not have fractures. There is considerable variability within and between families in age of onset and frequency of fractures Opens in new window.
During infancy and childhood, fracture frequency remains constant, but reduction in fracture frequency at puberty has been noted, followed by an increase in fracture frequency in women after menopause (22) or after the sixth decade in men.
Long bone deformity consists of bowing and angulation; however, it is not as severe in type I osteogenesis imperfecta as it can be in other forms.
About 20% of adults have kyphosis Opens in new window or scoliosis Opens in new window, which may be progressive. Trunk shortening has also been described.
Osteopenia Opens in new window may be minimal and undetectable on skeletal radiographs. In fact, radiography may fail to show any anomaly if pictures are taken during a period without a fracture. The ability to walk depends upon the type of osteogenesis imperfecta.
Peterson et al (21) found a significantly higher fracture rate in 5- to 20-year-old patients with type I osteogenesis imperfecta and opalescent teeth (type IB) than in those with type I who had normal teeth (type IA).
They also found that individuals with type IB were more likely to have had a fracture at birth, to have a higher fracture frequency, and to have a height below the second centile.
Patients with normal teeth were more than patients with opalescent teeth to have prolonged fracture-free periods during childhood. These two groups, however, were similar in frequency of joint hyperextensibility, bruising, hearing impairment, and joint dislocations.
Birth weights and birth lengths are generally normal. Short stature is of postnatal onset and usually mild; by adulthood, half the affected patients are less than the third centile for height.
Given the (usual) absence of significant skeletal deformity, the shortness of stature appears to be constitutional. However, individuals with the disorder whose height exceeds 6 feet have been noted.
Gillerot et al (23) reported a family with osteogenesis imperfect type I in which a newborn infant had fractures of type II. Heyes et al (24) reported a kindred with osteogenesis imperfecta type I and opalescent teeth; a stillborn had sustained multiple fractures in utero and had a membranous cranium, resembling that of osteogenesis imperfecta type II; no radiographs were depicted.
- Oral manifestations
Dental abnormalities have been described in many individuals with osteogenesis imperfecta; unfortunately, in many of these reports, the complete phenotype Opens in new window and inheritance pattern have not been described, and interpretation in view of current classification is not possible.
Heterogeneity based on the presence or absence of dental abnormalities has been noted (2). Paterson et al (21) recognized that two groups of families with osteogenesis imperfecta type I can be distinguished:
- a group with normal teeth (type IA) and
- a group with specific dental abnormalities (type IB).
In patients with dental abnormalities, deciduous and permanent teeth are opalescent, and amber or blue-gray on eruption.
On radiographic examination, there is increased constriction at the coronal-radicular junctions, and pulps become obliterated with secondary dentin. However, pulps may be wider than normal during early development (25).
Roots are thinner and shorter than normal. There is little variation in expression within the deciduous dentition, and these teeth may wear rapidly; on the other hand, there may be considerable variability in color within an individual’s permanent dentition.
Dental radiographs may be necessary to establish the diagnosis where tooth discoloration is mild.
When dental abnormalities are present, they may be helpful in making the diagnosis of osteogenesis imperfecta when the presence of other abnormalities is equivocal.
Lukinmaa et al, Paterson et al, and Lund et al noted that opalescent teeth were rarer in families with type I (8%) osteogeneis imperfecta than in those with type IV (37%). Malocclusion has been described and may be more common in individuals with opalescent teeth than in those with normal teeth.
Lukinmaa et al (26) described “thistle-tube” or flame-shaped pulps in several patients with osteogenesis imperfecta, one of whom may have had type I, although sclera color was not stated.
These dental findings are similar to those found in dentin dysplasia type II. Levin et al found similar abnormalities in the permanent dentitions of five members of a family with osteogenesis imperfecta type I.
There are few systematic studies of the histopathology of the teeth in what is unequivocally osteogenesis imperfecta type I. In general, however, the dentin-enamel junction has been reported by some investigators to be flatter than normal and to lack normal scalloping.
However, on scanning electron microscopy and light microscopy, others described a normal junction.
On light microscopic examination, laminated dentin, tubules of abnormal size and shape, and structures resembling entrapped blood vessels have been described.
Enamel surface and prism organization were found to be normal in osteogenesis imperfecta type IB. However, dentinal tubules were short, narrow, and tortuous compared with those of normal teeth from other osteogenesis imperfecta families and controls.
Gage et al studied teeth from a group of patients with a variety of osteogenesis imperfecta syndromes. They concluded that the majority of teeth from these patients were biochemically abnormal even if dental abnormalities were not present; however, the criteria for determining whether clinical abnormalities of the dentition existed in the patients studied were not presented.
- Other abnormalities
Easy bruisability has been found in about 75% of affected patients. Angiomatosis, hernias, and excessive sweating have also been reported. Nephrolitthiasis has been described.
See also:
- Clinical Features of Type II Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type III Osteogenesis ImperfectaOpens in new window
- Clinical Features of Type IV Osteogenesis ImperfectaOpens in new window
- Osteogenesis Imperfecta (Molecular Pathogenesis of the syndrome)Opens in new window
- Therapeutics for Patients with Osteogenesis ImperfectaOpens in new window
- Silence DO et al: Osteogenesis imperfecta. An expanding panorama of variants. Clin Orthoped Rel Res 159:11-25, 1981.
- Silence DO et al: Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 16:101-116, 1979.
- Silence DO et al: Clinical variability in osteogenesis imperfecta—variable expressivity or genetic heterogeneity. Birth Defects 15(5B): 113-129, 1979.
- Thompson EM et al: Recurrence risks and prognosis in severe sporadic osteogenesis imperfecta. J Med Genet 24:390-405, 1987.
- Bauze RJ et al: A new look at osteogenesis imperfect: A clinical, radiological and biochemical study of forty-two patients. J Bone Joint Surg Br 57:2-12, 1975.
- Spranger J: Osteogenesis imperfect: A pasture for splitters and lumpers. Am J Med Genet 17:425-428, 1984.
- Stoss H et al: Heterogeneity of osteogenesis imperfect. Biochemical and morphological findings in a case of type III according to Sillence. Eur J Pediatr 145:34-39, 1986.
- Stoll C et al: Birth prevalence rates of skeletal dysplasias. Clin Genet 35:88-92, 1989.
- Smith R et al: The eye and collagen in osteogenesis imperfecta. Birth Defects 12(3):563-566, 1976.
- Sillence DO et al: Natural history of blue sclera in osteogenesis imperfecta. Am J Med Genet 45:183-186, 1993.
- Smith R et al: The Brittle Bone Syndrome. Osteogenesis Imperfecta. Butterworths, London, 1983.
- Carey MC et al: Osteogenesis imperfecta in twenty-three members of a kindred with heritable features contributed by a nonspecific skeletal disorder. Q J Med 37:437-449, 1968.
- Madigan WP et al: Retinal detachment in osteogenesis imperfecta. J Pediatr Ophthalmol Strabismus 31:268-269, 1994.
- Carpenter BF, Hunter AGW: Micromelia, polysyndactyly, multiple malformations, and fragile bones in a stillborn child. J Med Genet 19:311, 1982.
- Reidner ED et al: Hearing patterns in dominant osteogenesis imperfecta. Arch Otolaryngol 106:737-740, 1980.
- Cox JR, Simmons CL: Osteogenesis imperfecta and associated hearing loss in five kindreds. South Med J 75:1222-1226, 1982.
- Garretsen TJTM, Cremers CWRJ: Clinical and genetic aspects in autosomal dominant inherited osteogenesis imperfect type 1. Ann N Y Acad Sci 630:240-248, 1991.
- Hortop J et al: Cardiovascular involvement in osteogenesis imperfecta. Circulation 73:54-61. 1986.
- Pyeritz RE, Levin LS: Aortic root dialatation and valvular dysfunction in osteogenesis imperfecta. Circulation 64:1193A, 1981.
- White NJ et al: Cardiovascular abnormalities in osteogenesis imperfect. Am Heart J 106:1416-1420, 1983.
- Paterson CR et al: Osteogenesis imperfecta with dominant inheritance and normal sclerae. J Bone Joint Surg Br 65:35-39, 1983.
- Paterson CR et al: Osteogenesis imperfect after the menopause. N Engl J Med 310:1694-1696, 1984.
- Gillerot Y et al: Lethal perinatal type II osteogenesis imperfect in a family with a dominantly inherited type I. Eur J Pediatr 141:119-122, 1983.
- Heyes FM et al: Osteogenesis imperfecta and odontogenesis imperfecta: Clinical and genetic aspects in eighteen families. J Pediatr 56:234-245, 1960.
- Levin LS et al: The dentition in the osteogenesis imperfecta syndromes. Clin Orthop Rel Res 159:64-74, 1981.
- Lukinmaa P-L et al: Dental findings in osteogenesis imperfecta I. occurrence and expression of type I dentinogenesis imperfecta. J Craniofac Genet Dev Biol 7:115-125, 1987.
- Pozo JL et al: Basilar impression in osteogenesis imperfecta. A report of three cases in one family. J Bone Joint Surg Br 66:233-238, 1984.
