
Fragile X Syndrome
 File Photo. Credit: Health JadeOpens in new window
File Photo. Credit: Health JadeOpens in new window
|
Fragile X syndrome is a genetic condition that causes a range of developmental problems including learning disabilities and cognitive impairment. Usually, males are more severly affected by this disorder than females.
Affected individuals usually have delayed development of speech and language by age 2.
Most males with fragile X syndrome have mild to moderate intellectual disability, while about one-third of affected females are intellectually disabled.
Children with fragile X syndrome may also have anxiety and hyperactive behavior such as fidgeting or impulsive actions. They may have attention deficit disorder, which includes an impaired ability to maintain attention and difficulty focusing on specific tasks.
About one-third of individuals with fragile X syndrome have features of autism spectrum disorder that affect communication and social interaction. Seizures occur in about 15 percent of males and about 5 percent of females with fragile X syndrome.
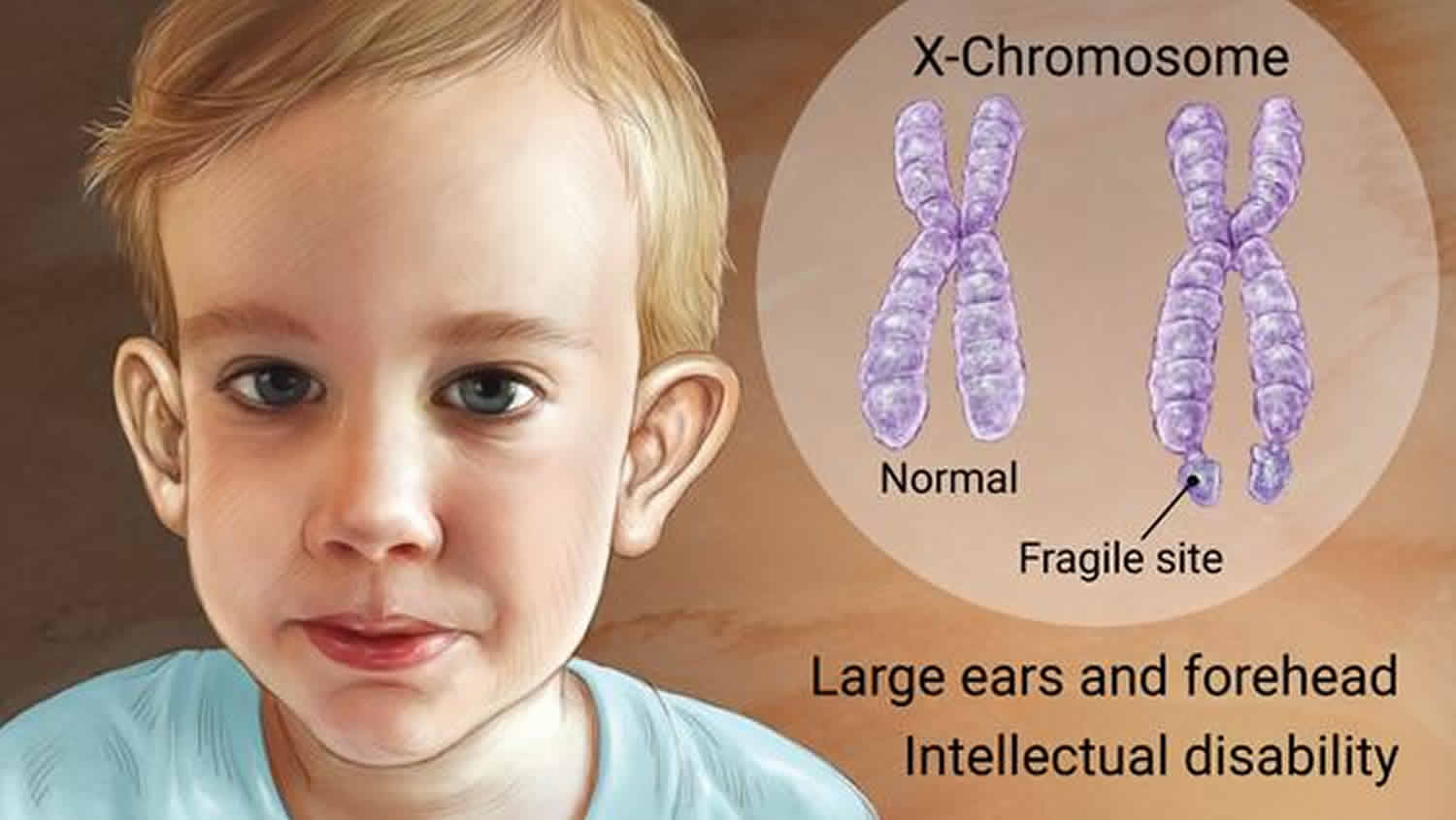
Most males and about half of females with fragile X syndrome have characteristic physical features that become more apparent with age. These features include a logn and narrow face, large ears, a prominent jaw and forehead, unusually flexible fingers, flat feet, and in males, enlarged testicles (macroochidism) after puberty.
Causes of Fragile X Syndrome
Mutations in the FMR1 gene cause fragile X syndrome. The FMR1 gene provides instructions for making a protein called “FMRP.” This protein helps regulate the production of other proteins and plays a role in the development of synapses, which are specialized connections between nerve cells. Synapses are critical for relaying nerve impulses.
Nearly all cases of fragile X syndrome are caused by a mutation in which a DNA segment, known as the “CGG triplet repeat,” is expanded within the FMR1 gene. Normally, this DNA segment is repeated from 5 to about 40 times.
In people with fragile X syndrome, however, the CGG segment is repeated more than 200 times. The abnormally expanded CGG segment turns off (silences) the FMR1 gene, which prevents the gene from producing FMRP. Loss or a shortage (deficiency) of this protein disrupts nervous system functions and leads to the signs and symptoms of fragile X syndrome.
Males and females with 55 to 200 repeats of the CGG segment are said to have an FMR1 gene permutation. Most people with this permutation are intellectually normal. In some cases, however, individuals with a permutation have lower than normal amounts of FMRP. As a result, they may have mild versions of the physical features seen in fragile X syndnrome (such as prominent ears) and may experience emotional problems such as anxiety or depression.
Some children with an FMR1 premutation may have learning disabilities or autistic-like behavior. The permutation is also associated with an increased risk of disorders called “fragile X-associated primary ovarian insufficiency” (FXPOI) and “fragile X-associated tremor/ataxia syndrome” (FXTAS).
Inheritance of Fragile X Syndrome
Fragile X syndrome is inherited in an X-linked dominant pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. (The Y chromosome is the other sex chromosome.) The inheritance is dominant if one copy of the altered gene in each cell is sufficient to cause the condition.
X-linked dominant means that in females (who have two X chromosomes), a mutation in one of the two copies of a gene in neach cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of a gene in each cell causes the disorder. In most cases, males experience more severe symptoms of the disorder than females.
In women, the FMR1 gene permutation on the X chromosome can expand to more than 200 CGG repeats in cells that develop into eggs. This means that women with the permutation have an increased risk of having a child with fragile X syndrome. By contrast, the permutation in men does not expand to more than 200 repeats as it is passed to the next generation. Men pass the permutation only to their daughters. Their sons receive a Y chromosome, which does not include the FMR1 gene.
- Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 1993;91(2):398 – 402.
- Gunay-Aygun M, Schwartz S, Heeger S, et al. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001; 108(5):E92.
- Haqq Am, Grambow SC, Muehlbauer M, et al. Ghrelin concentrations in Prader-Willi syndrome (PWS) infants and children: changes during development. Clin Endocrinol (Oxf) 2008;69(6):911 – 20.
