
Goltz Syndrome (Focal Dermal Hypoplasia)
Clinical Definition and Overview
|
Goltz syndrome (also called Focal dermal hypoplasia), an uncommon X-linked dominant condition first described by Goltz in 1962, is a disease of mesoectodermal development caused by mutations in the PORCN gene, which affects ectodermal and mesodermal structures — primarily the skin, nails, hair, bones, eyes and teeth— in a mosaic pattern. |
Specifically, patients exhibit pigmentary and atrophic skin changes, nail dystrophy, thin hair, syndactyly, bone hypoplasia, and small teeth with dental hypoplasia. Over 80% of affected individuals are female, suggesting that the disorder is lethal in male hemizygotes.
Genetics
The mosaic distribution of skin lesions in female patients with Goltz syndrome reflects lyonization Opens in new window.
Although Goltz syndrome is antenatally lethal in boys with non-mosaic hemizygous mutations (as in incontinentia pigmenti Opens in new window), male patients with genomic mosaicism (e.g. due to a postzygotic mutation) or functional (in the setting of Klinefelter syndrome Opens in new window) account for 10% of affected individuals.
Approximately 95% of all Goltz syndrome case are sporadic, which likely reflects both a decreased likelihood of reproduction by severely affected women and the lethality of PORCN mutations with widespread expression.
Some authors have postulated that either preferential inactivation of the mutant X chromosome Opens in new window or a postzygotic mutation is required for survival of affected female fetuses. Patients with the most deleterious mutations (deletions) have more extreme skewing in favor of wild-type.
Rare familial cases show anticipation Opens in new window, with the offspring being much more severely affected than the parent and having a higher proportion of mutant cells. Therefore the possibility that an apparently normal mother is a carrier must be considered when counseling families.
Pathogenetics
PORCN belongs to the evolutionarily conserved porcupine gene family and is expressed in the skin, craniofacial and long bones, tooth buds and eyes—structures affected in Goltz syndrome.
The PORCN protein has multiple transmembrane domains and is located in the endoplasmic reticulum. It functions as an O-acyltransferase involved in palmitoylation and secretion of Wnt, a morphogen important in ectomesodermal tissue development.
Wnt Opens in new window signaling stimulates fibroblast proliferation, inhibits adipogenesis and induces osteogenesis, explaining the dermal hypoplasia, fat “herniations” and osteopathia striata (streaks of decreased bone density) observed in Goltz syndrome. Wnt also has key roles in early limb patterning and tooth formation.
Although the primary cutaneous abnormality in Goltz syndrome is in the dermis, the distribution of skin lesions along Blaschko’s lines Opens in new window reflects embryonic migration pathways of epidermal cells.
Ectoderm-specific deletion of PORCN in mice has shown that expression of this gene in the epidermis and resultant Wnt signaling regulate development of the underlying dermis and fat.
Of note, patients with Goltz syndrome have a thin epidermis (as well as dermis) and abnormal adnexal structures. This is not surprising considering that Wnt signaling, including β-catenin-dependent pathways, is known to have critical roles in epidermal regeneration and adnexal morphogenesis/maintenance.
In 3 of 24 patients in one series, a microdeletion affected both the PORCN gene and the adjacent EBP gene responsible for Conradi-Hünerman-Happle syndrome Opens in new window.
Curiously, however, these patients showed no features of the latter disorder. This implies that the PORCN hypoplastic skin phenotype predominates over the ichthyosis and follicular changes of Conradi-Hünerman-Happle syndrome Opens in new window.
Clinical Features
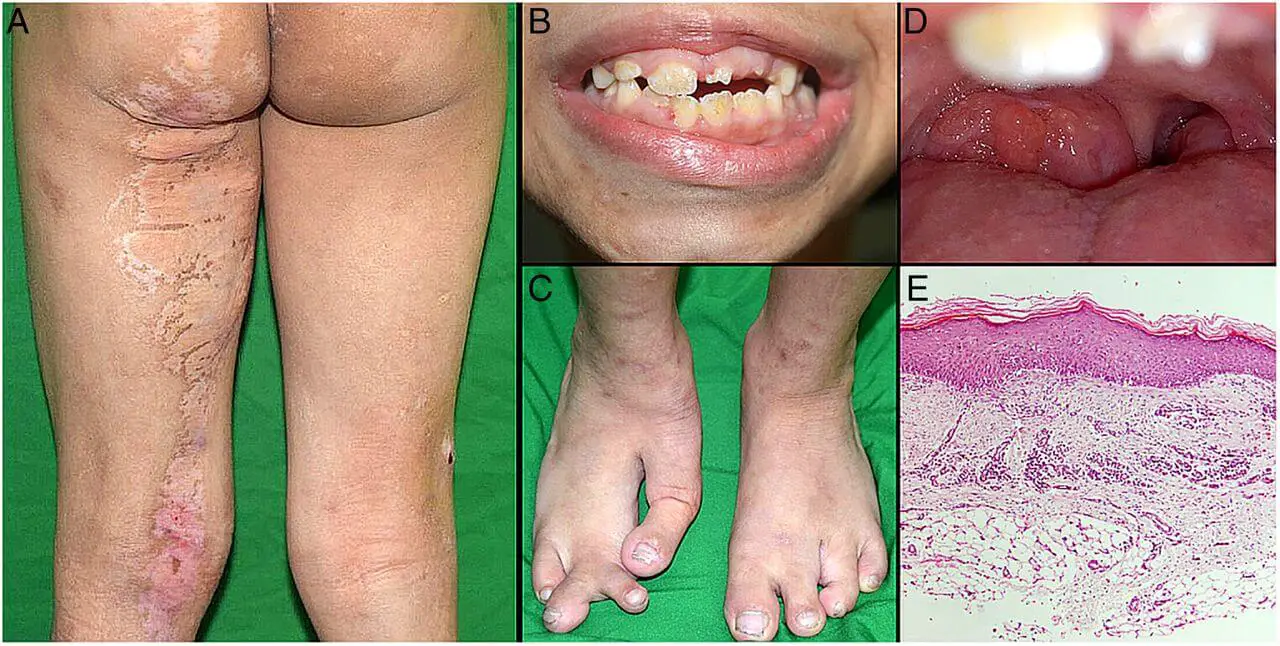
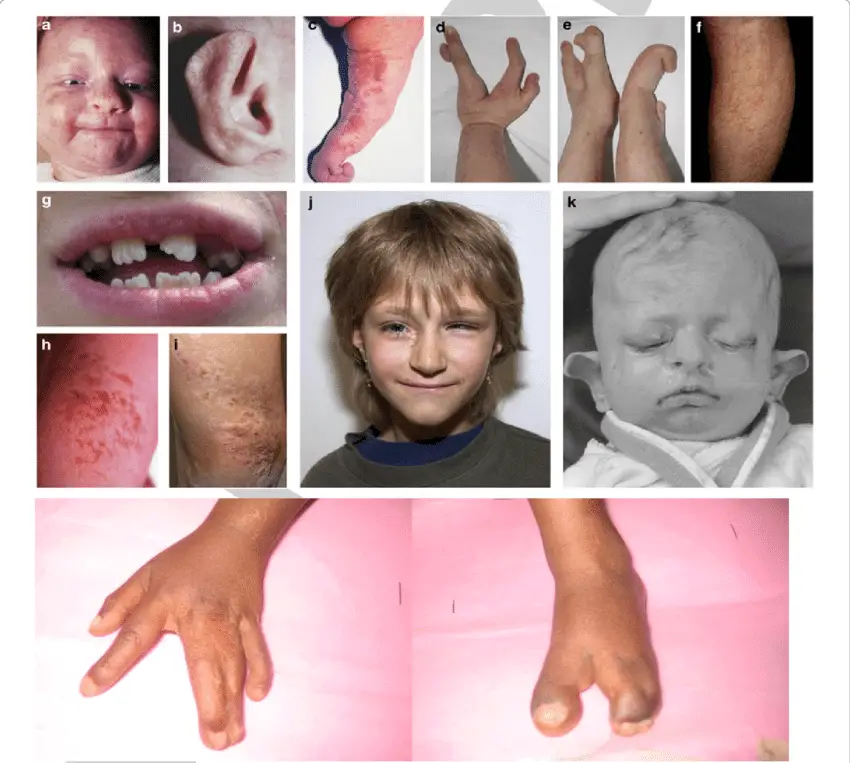
The phenotype Opens in new window of Goltz syndrome varies depending on the proportion and distribution of cells expressing a mutant X chromosome. Streaks of vermiculate dermal atrophy and/or telangiectasias are often present from birth. Later, hypo- and hyperpigmentation as well as outpouchings of fat develop.
|
Although raspberry-like papillomas may appear in any location, they favor the anogenital region as well as the lips, larynx and acral sites. Bony reduction deformities of the hands are common, particularly ectrodactyly (split-hand/foot or “lobster-claw” malformation).
Radiographs of the mid portion of the lower extremities characteristically show osteopathia striata. Eye abnormalities, often unilateral, include coloboma, aniridia, microphthalmia and anophthalmia.
Dystrophic nails (e.g. longitudinal fissures, hypoplasia), sparse hair, abnormal teeth (e.g. notched nasal alae, pointed chin, large malformed ears) may be observed.
Other occasional features include aplasia cutis congenital, myelomeningocele, cleft lip/palate, deafness, urinary tract anomalies and gastrointestinal maldevelopment.
Pathology
Affected skin shows a reduction in dermal collagen, which can be marked with telangiectasias and a decreased number of adnexal structure. Fat cells of varying sizes are often present in the upper dermis.
By electron microscopy, the basement membrane zone can be seen to contain multiple layers of lamina densa-like material, a change consistent with keratinocyte damage. Dermal microfibrils are also increased in the upper dermis.
Differential Diagnosis
The distinctive cutaneous and extracutaneous findings of Goltz syndrome, especially dermal atrophy/fat “herniations” and osteopathia striata, distinguish it from other X-linked dominant genodermatoses.
Examples of such conditions include MIDAS Opens in new window (which does not involve the distal extremities), Conradi-Hünerman-Happle syndrome Opens in new window and oral-facial-digital type 1 syndromes Opens in new window. Although an inflammatory component is occasionally observed, the skin lesions of Goltz syndrome are relatively static in comparison to the evolving lesions of IP.
Treatment
Treatment is supportive, with appropriate subspecialist referral based on the associated abnormalities. The telangiectasias may be improved by pulsed dye laser treatment. Troublesome exophytic papillomas can be treated with curettage or photodynamic therapy.
Corticosteroids, adrenocorticotropic hormone (ACTH), and vigabatrin have been used for the treatment of infantile spasms. Motor disturbances may be minimized with good physiotherapy and orthopedic care.
Mental retardation is approached educationally rather than medically, and training must be appropriate to the patient’s individual capabilities.
Ocular, oral, urogenital, and other disturbances must receive appropriate individual treatment. No special treatment is indicated for the cutaneous lesions.
See also:
- Bellosta M, Trespiolli D, Ghiselli E, et al. Focal dermal hypoplasia: report of a family with 7 affected women in 3 generations. Eur J Dermatol 1996;6:499-500.
- Goltz RW. Focal dermal hypoplasia syndrome: an update (Editorial). Arch Dermatol 1992;128:1108-1111.
- Holzman RS. Airway involvement and anesthetic management in Goltz ‘s syndrome. J Clin Anesth 1991;3:422-425.
