
Hereditary Hemorrhagic Telangiectasia
Clinical Features, Genes, and Therapeutics
Hereditary hemorrhagic telangiectasia (HHT) (also called Osler-Weber-Rendu syndrome) is an inherited autosomal dominant disorder of the blood vessel walls resulting in telangiectasias. Blood vessels in this disease are unusually thin due to inadequate elastic tissue and smooth muscle, leading to vessel dilatation.
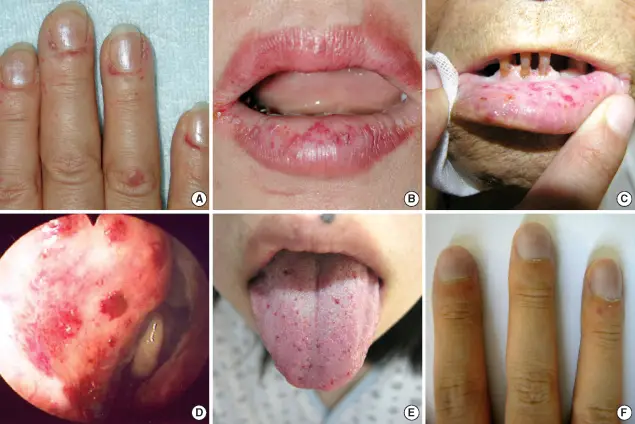
|
| Clinical presentation courtesy of ResearchGate Opens in new window | Clinical features of the HHT patients: (A) telangiectasia on the periungal regions of the fingers of patient 1, (B) telangiectasia on the lips of patient 1, (C) telangiectasia on the oral mucosa of the father of patient 2, (D) bleeding foci in the Kisselbach's plexus of patient 3; (E) microtelangia on the tongue of patient 5, and (F) clubbing fingers with one small hemangioma in patient 5. |
Patients with HHT manifest clinical symptoms characterized by punctuate telangiectasias located on the lips, face, palms/soles, nail beds, tongue, nasopharynx, and the gastrointestinal and genitourinary tracts. Recurrent epistaxis and visceral arteriovenous malformations (AVMs), especially in the lung, liver, brain, and bowel, are common entities of this hereditary disorder.
Epidemiologic studies suggest a prevalence that ranges from 1 in 5,000 to 18,000 people, depending on the geographic region. Mutation Opens in new window of either one of two different genes at two separate loci is responsible for the condition.
HHT1 is caused by a mutation of the endoglin (ENG) gene on chromosome 9, whereas mutation of activin receptor-like kinase-1 (ALK1; ACVRLI), a gene located on chromosome 12, produces HHT2.
The proteins produced by these genes Opens in new window may play a role in blood vessel wall integrity. With both types of HHT, numerous vascular hamartomas develop, affecting the skin and mucosa; however, other vascular problems, such as arteriovenous fistulas, may also be seen.
Patients with HHT1 tend to have more pulmonary and cerebral involvement, whereas those with HHT2 generally have a later onset of their telangiectasias and a greater degree of hepatic involvement. A much less common mutation, involving the MADH4 gene, has also been identified, and these patients exhibit an overlap syndrome characterized by HHT and juvenile polyposis.
The polyps involve both the upper and lower gastrointestinal tract, and these patients have an increased risk for developing colorectal carcinoma at an early age. The clinician should be familiar with HHT because the oral lesions are often the most dramatic and most easily identified component of this syndrome.
Clinical Features
Patients with HHT are often diagnosed initially because of frequent episodes of epistaxis.
On further examination, the nasal and oropharyngeal mucosae exhibit numerous scattered red papules, 1 to 2 mm in size, which blanch when diascopy is used.
This blanching indicates that the red color is due to blood contained within blood vessels (in this case, small collections of dilated capillaries [talangiectasias] that are close to the surface of the mucosa).
These telangiectatic vessels are most frequently found on the vermilion zone of the lips, tongue, and buccal mucosa, although any oral mucosal site may be affected. With aging, the telangiectasias tend to become more numerous and slightly larger.
In many patients, telangiectasias are seen on the hands and feet. The lesions are often distributed throughout the gastrointestinal mucosa, the genitourinary mucosa, and the conjunctival mucosa.
The gastrointestinal telangiectasias have a tendency to rupture, which may cause significant blood loss. Chronic iron-deficiency anemia is often a problem for such individuals. Significantly, arteriovenous fistulas may develop in the lungs (15% to 45% of HHT patients), liver (30%), or brain (10% to 20%).
The pulmonary arteriovenous malformations seem to predispose these patients to the development of brain abscesses due to right-to-left shunting of bacteria that might be introduced into the bloodstream.
In at least one instance, periodontal vascular malformations were left to be the cause of septic pulmonary emboli that resolved only after several teeth with periodontal abscesses were extracted.
A diagnosis of HHT can be made if a patient has three of the following four criteria:
- Recurrent spontaneous epistaxis
- Telangiectasias of the mucosa and skin
- Arteriovenous malformation involving the lungs, liver or central nervous system (CNS)
- Family history of HHT
In some instances, CREST syndrome Opens in new window (Calcinosis cutis, Raynaud phenomenon, Esophageal dysfunction, Sclerodactyly, and Telangiectasia) must be considered in the differential diagnosis. In these cases, serologic studies for anticentromere autoantibodies often help to distinguish between the two conditions because these antibodies typically would be present only in CREST syndrome.
Histopathologic Features
If one of the telangiectasias is submitted for biopsy, the microscopic features essentially show a superficially located collection of thin-walled vascular spaces that contain erythrocytes.
Treatment and Prognosis
For mild cases of HHT, no treatment may be required. Moderate cases may be managed by selective cryosurgery or electrocautery of the most bothersome of the telangiectatic vessels.
Later ablation of the telangiectatic lesions has also been used, although this approach appears to be most successful for patients with mild to moderate disease.
More severely affected patients, particularly those troubled by repeated episodes of epistaxis, may require a surgical procedure of the nasal septum (septal dermoplasty).
The involved nasal mucosa is removed and replaced by a skin graft; however, some long-term follow-up studies suggest that the grafts eventually become revascularized, resulting in recurrence of the problem. Nasal closure is another surgical technique that has been performed for patients with severe epistaxis in whom other methods have failed.
Combined progesterone and estrogen therapy may benefit some patients, but because of the potentially serious side effects, this should be limited to the most severely affected individuals.
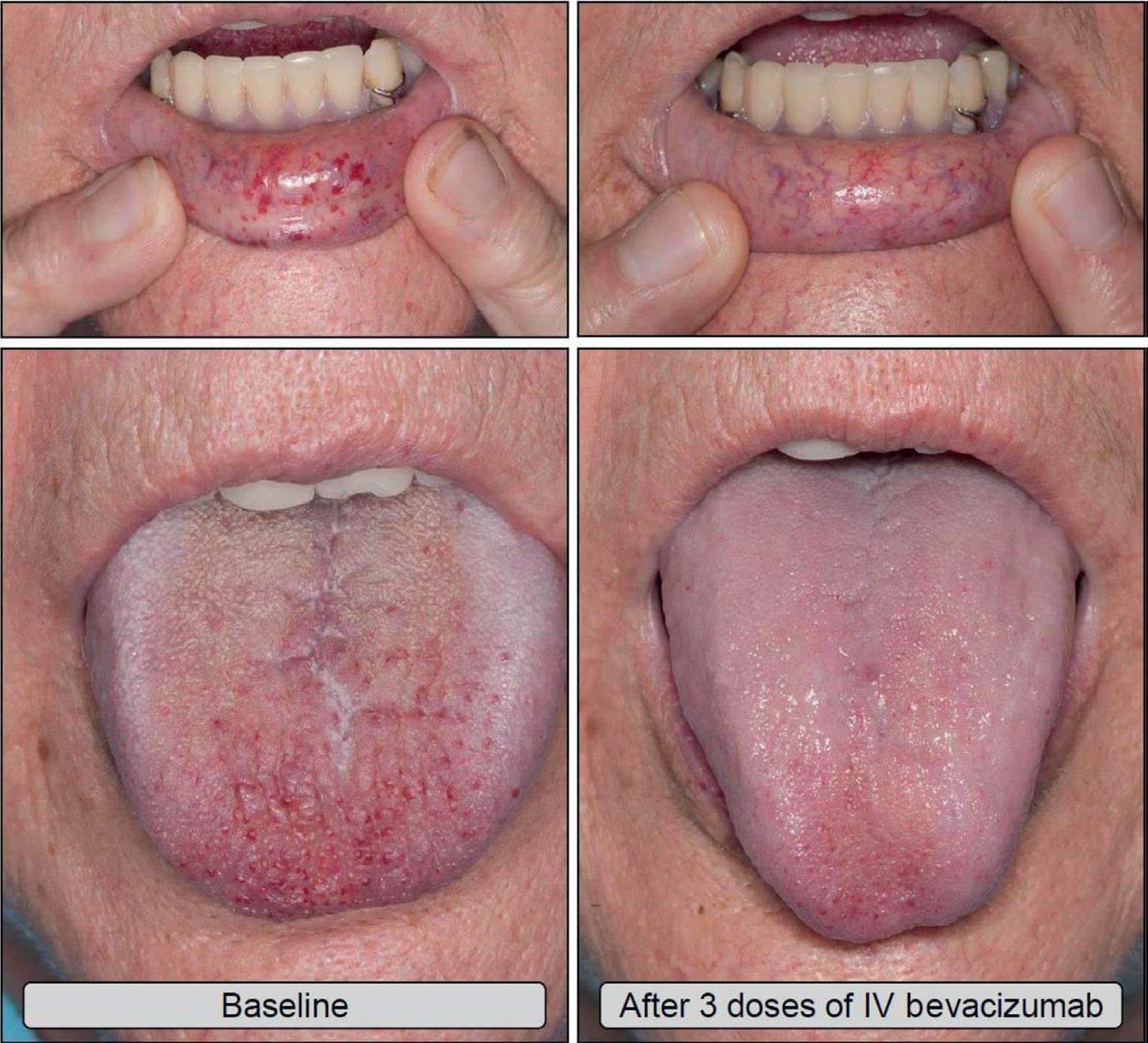
|
Bevacizumab, an antibody directed against vascular endothelial growth factor, has shown some promise in controlling epistaxis, but this is a costly medication. Iron replacement therapy is indicated for the iron-deficit patient, and occasionally blood transfusions may be necessary to compensate for blood loss.
From a dental standpoint, some authors recommend the use of prophylactic antibiotics before dental procedures that might cause bacteremia in patients with HHT and evidence of a pulmonary arteriovenous malformation.
For patients with a history of HHT, such antibiotics are advocated until a pulmonary arteriovenous malformation is ruled out because of the 1% prevalence of brain abscesses in affected individuals.
Patients with a history of HHT should be screened for arteriovenous malformations, which can be eliminated by embolization or other vasodestructive techniques using interventional radiologic methods. The decision to treat such a lesion often depends on the anatomic site and the severity of the malformation.
