
Conradi-Hünermann-Happle Syndrome
Clinical Definition and Features
|
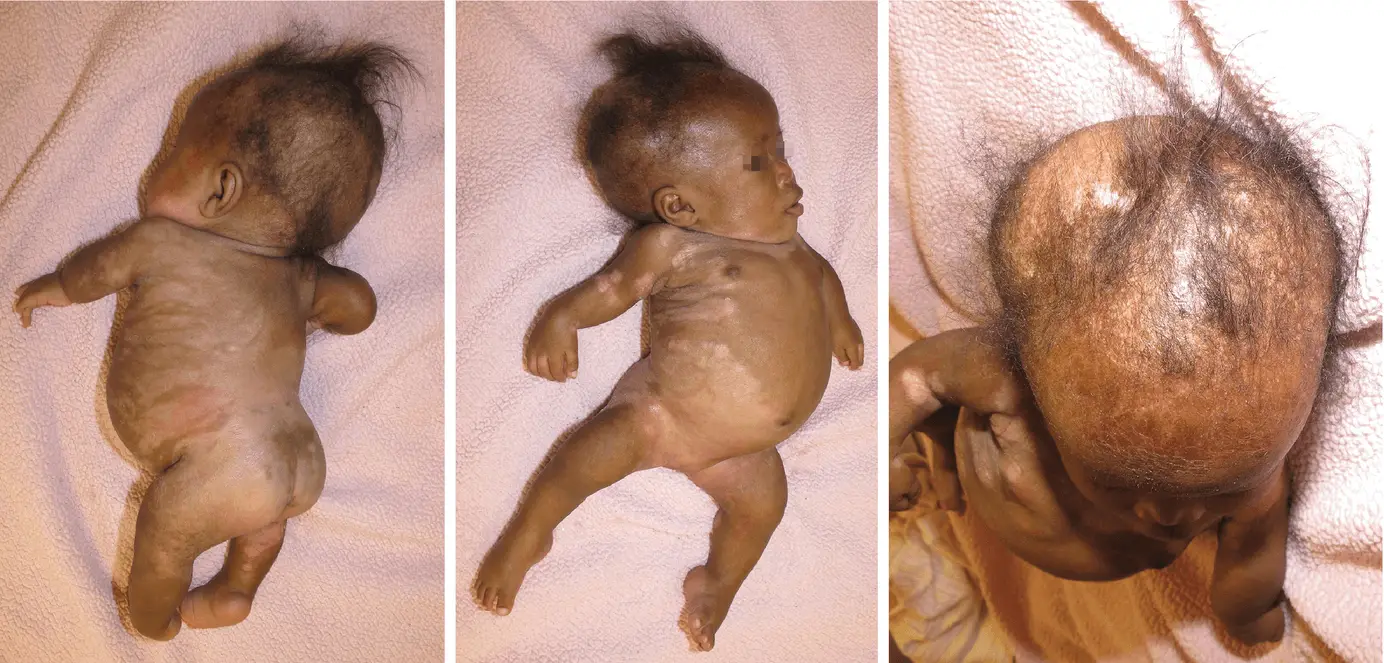
Conradi-Hünermann-Happle syndrome is a multisystem disorder involving skin, bones and eyes. CHH is an exceedingly rare X-linked dominant trait Opens in new window which exclusively affects female neonates with ichthyosiform erythroderma characterized by linear ichthyosis Opens in new window, chondrodysplasia punctata Opens in new window, cataracts Opens in new window, and short stature Opens in new window. Thus, the syndrome is also often called X-linked dominant chondrodysplasia punctata Opens in new window. |
Extracutaneous findings include rhizomelic limb Opens in new window shortening with epiphyseal stippling (chondrodysplasia punctata) and ocular anomalies such as sectorial cataracts and microphthalmia. Patients often have swirled alopecia Opens in new window following Blaschko’s lines Opens in new window and coarse, lusterless hair. After infancy, the skin changes evolve into follicular atrophoderma.
CHH syndrome is caused by mutations Opens in new window in the emopamilbinding protein (EBP) gene, which lies on the X chromosome Opens in new window adjacent to the PORCN gene and encodes an isomerase essential for cholesterol biosynthesis. Of note, chondrodysplasia punctata occurs in a variety of other genetic and non-genetic disorders.
Cutaneous (Skin) Involvement
At birth, the skin of neonates often exhibits severe ichthyosiform erythroderma and the affected girls are born with yellowish markedly hyperkeratotic Opens in new window plaques.
After the first 3 to 6 months of life the erythroderma and scaling resolve, leaving erythema and later follicular atrophoderma Opens in new window in a distribution that follows Blaschko lines, hypopigmentation and hyperpigmented streaks particularly on the trunk, and circumscribed cicatricial alopecia of the scalp and eyebrows triginous areas (ptychotropism). However, ichthyosis on arms and legs often remains present during the entire life.
Many patients show minor nail anomalies, e.g., flattening of the nail plates (platnychia) or splitting into layers (onychoschisis). Particularly, in adult patients, patchy areas of cicatricial alopecia are a constant feature and sometimes may even be the only skin manifestation seen in mildly affected females.
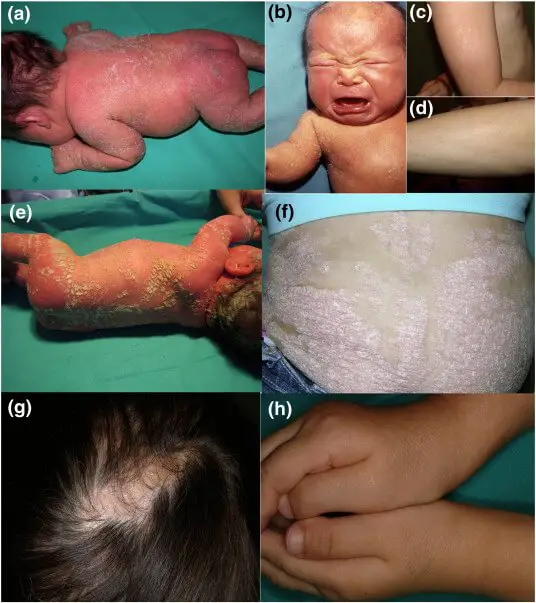
|
| Figure X-2. Clinical features of CHH syndrome | Photo courtesy of ScienceDirect Opens in new window |
Non-Cutaneous Findings
A hallmark of the CHH syndrome is the asymmetry of skeletal involvement. Patients with CHH exhibit asymmetric skeletal involvement with punctuate calcification of the epiphyseal regions that usually results in an asymmetric shortening of the long bones (especially the humeri and femora) and sometimes in severe kyphoscoliosis, facial dysplasia, and congenital hip dislocation.
Unilateral or bilateral sectorial cataracts; patchy, coarse, lusterless hair, nasal bone dysplasia with saddlenose deformity, and a high-arched palate further characterize the signs of the disease.
The bony abnormalities but not the skin lesions of CHH syndrome have been described in association with teratogenic exposure to medications and infections and maternal autoimmune diseases.
Biochemical and Genetic Description
Happle delineated the CHH syndrome as a distinct type of chondrodysplasia punctata Opens in new window characterized by a mosaic pattern of skin lesions. As in incontinentia pigmenti Opens in new window, the disease occurred primarily in females and the cutaneous changes showed a similar linear distribution pattern with widespread atrophic lesions, pigmentary disturbances and ichthyosis.
Based on these peculiar cutaneous findings Happle argued that he was dealing with a further X-linked dominant gene defect that is usually lethal in males, and hypothesized that the linear cutaneous involvement was an example of X-chromosome inactivation Opens in new window and functional mosaicism.
A peculiar phenomenon seen in the disease is anticipation, a stepwise increase in disease expression from one generation to the next. In part, this anticipation may reflect the presence of gonadal and somatic mosaicism in the parental generation.
Methylation patterns of the X-chromosome Opens in new window can show extreme skewing so that the disease can manifest mildly in a patient, but can be extreme in the child as an alternative explanation for anticipation.
Mutations Opens in new window have been identified in the gene that encodes emopamil binding protein (EBP). The EBP gene Opens in new window resides on the short arm of the X-chromosome at Xp11.22-p11.23 and the mouse homolog is mutated in the mouse mutant tattered (Td).
The EBP gene has a dual function: on the one it serves as a binding protein for the calcium antagonist emopamil and thus as a highly affinity receptor for several anti-ischemic drugs, and on the other hand it also acts as a δ8 – δ7 sterol isomerase, which is a key enzyme in the final steps of cholesterol biosynthesis.
Due to the enzymatic block cholesterol precursor molecules such as 8-dehydrocholestero (8-DHC) and cholest-8(9)-en-3-beta-ol are markedly accumulated, which allows a rapid lipid-biochemical diagnosis of the CHH syndrome by gas chromatography-mass spectrometry (GC-MS) studies.
However, the extent of the metabolic alterations does not correlate well with the molecular genotype, nor with the severity of the clinical phenotype. This lack of correlation is probably due to differences in X-inactivation Opens in new window between different tissues of the same patient.
The severe disturbance of cholesterol biosynthesis seen in this disorder is certainly also the metabolic cause for the severe congenital skeletal dysplasia.
Other metabolic defects of cholesterol biosynthesis, such as the Smith-Lemli-Opitz syndrome Opens in new window or the CHILD syndrome Opens in new window, are also associated with congenital skeletal dysplasia and with chondrodysplasia punctata.
A lesson that can be learned from the CHH syndrome is that vertebrate embryonic development is dependent on large amounts of cholesterol which is needed for growth and for the synthesis of compounds derived from cholesterol, such as steroid hormones.
An abnormal cholesterol metabolism obviously impairs the function of sonic hedgehog and other related embryonic signaling proteins that help determine the vertebrate body plan during the earliest weeks of embryonic development.
See also:
- Happle R. X-linked dominant chondrodysplasia punctata. Review of literature and report of a case. Hum Genet. 1979;53:65-73.
- Happle R. Genetic interpretation of linear skin abnormalities. Hautzart. 1978;29:357-363.
- Traupe H, Müller D, Atherton D, et al. Exclusion mapping of the X-linked dominant chondrodysplasia punctata/ichthyosis/cataract/short stature (Happle) syndrome: possible involvement of an unstable premutation. Hum Genet 1992;89:659-665.
- Braverman N, Lin P, Mocbius FE, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat Genet. 1999;22:291-294.
- Kelly RI, Wilcox WG, Smith M, et al. Abnormal sterol metabolism in patients with Conradi-Hünermann-Happle syndrome and sporadic lethal chondrodysplasia punctata. Am J Med Genet. 1999;83:213-219.
